得られるデータ
MA-plot, デンドログラム, ヒートマップ, 遺伝子オントロジー, Fastqファイル, カウント一覧・FPKM一覧, BAMファイル, BedGraphファイル等
CAGE-seqとは
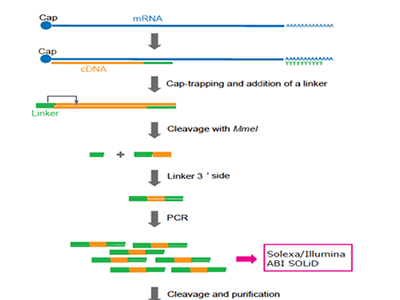
CAGE-seqは、理化学研究所にて開発されたRNAseqやマイクロアレイに代わる新しい発現解析方法で、完全長のcDNAのみを回収し、mRNAやlncRNAの発現量および転写開始点(Transcription starting site)を網羅的に解析する方法です。その特徴は、RNAの5’末端に付加されるキャップ構造を特異的にビオチン化して回収するところにあります(Cap-trap法)。PCRによる増幅工程をライブラリー作製過程から完全に排除することにより、CAGE-seqは、他の方法よりも定量性に優れ、広いダイナミックレンジを有しています。さらに、転写因子結合モチーフやalternative promoterの探索、高度な遺伝子制御ネットワークの推定など、CAGE-seqの結果からは多くの情報を得ることができます。
サービスの特徴
・各遺伝子を転写開始点ごとに定量:世界で唯一、ゲノムワイドにプロモーター活性を定量解析できる技術
・定量性に優れる:PCRの工程が無く、5’末端に特化してシークエンスするため、RNA-seqのように長さのバイアスも無く、定量性に優れる
・高感度:200万のRNA分子から1つのRNAを99.99%の確度で検出可能
・新規RNA分子(mRNA、lncRNAやエンハンサーRNAなどのncRNA)・プロモーターを発見できる可能性もある
・転写因子結合モチーフ探索:発現変動が確認された転写開始点周辺情報を基に、モチーフ配列を探索することで、関与していた可能性のある転写因子を予測することができます
・大規模、かつ、高速の解析が低コストで実現可能
・理研とダナフォームが共同開発した技術